Analyzing My Dog's DNA
January 21, 2019 • 🐕🧬
Dogs are awesome. Since March of 2014, I've owned a little dog named Riker:

I adopted her from the Humane Society in Milpitas. She was 8 weeks old. She recently had her fifth birthday. Happy birthday Riker! I love her very much.
Anyways, when I got her, I was curious about what type of dog she was. She looks like a chihuahua, but has some features that don't match the traditional chihuahua:
- Curled, fluffy tail
- Gradient-banded coat near her front legs
- Sheds mountains of fur
- Generally pleasant demeanor
I thought she might have been part Shiba Inu. These dogs are adorable and have the curled tail and gradient-banded coat. Here's one from Wikipedia:
A shiba inu dog outside on grass
{kind=link}
Doesn't this dog look like Riker? With the tail and banded coat? I thought so too.
Whenever other people asked me about Riker, I would tell them she's "part chihuahua, part shiba inu". They believed me, but I didn't have any proof.
Enter Embark
A good friend of mine told me about a service where you can get your dog's DNA analyzed. It's called EmbarkVet (disclaimer: this is a referral link). Embark gives your dog a breed report, health analysis, and some cool genetic information. It's like 23andMe for your dog.
I signed up for the service. A few days later they sent me a DNA swab kit. It's like an oversized q-tip. After sticking this in Riker's cheeks for 30 seconds or so (she wasn't happy about this, but I told her it was for science), I packed the kit in the return shipping and sent it off.
Over the next few weeks, Embark notified me as the analysis progressed. After 3 weeks or so, my pressing question was answered: they told me what type of dog she was!
Less than 50% chihuahua
To my absolute amazement, Riker is not even mostly chihuahua! The results confirm that Riker is a pomeranian! The full breakdown is:
- 50% Pomeranian (this accounts for the curved tail, banded coat, shedding, and how she's nice to be around)
- 32.5% Chihuahua
- 6.9% Miniature Pinscher (no relation to the larger dog)
- 10.6% "Supermutt" (small amounts of DNA from other dogs, including Cocker Spaniel)
I also learned that she has 1.5% "Wolfiness", which indicates ancient wolf genes that have survived through to modern domesticated dogs. Cool!
Note: I am not a trained biologist. Regrettably, I spent much of my college biology course playing Pokémon Crystal, although I do remember some parts of the genetics sections.
A family tree
Embark also determines haplotypes, which means they can tell what genes of Riker's came from which parents. This lets them generate a family tree:
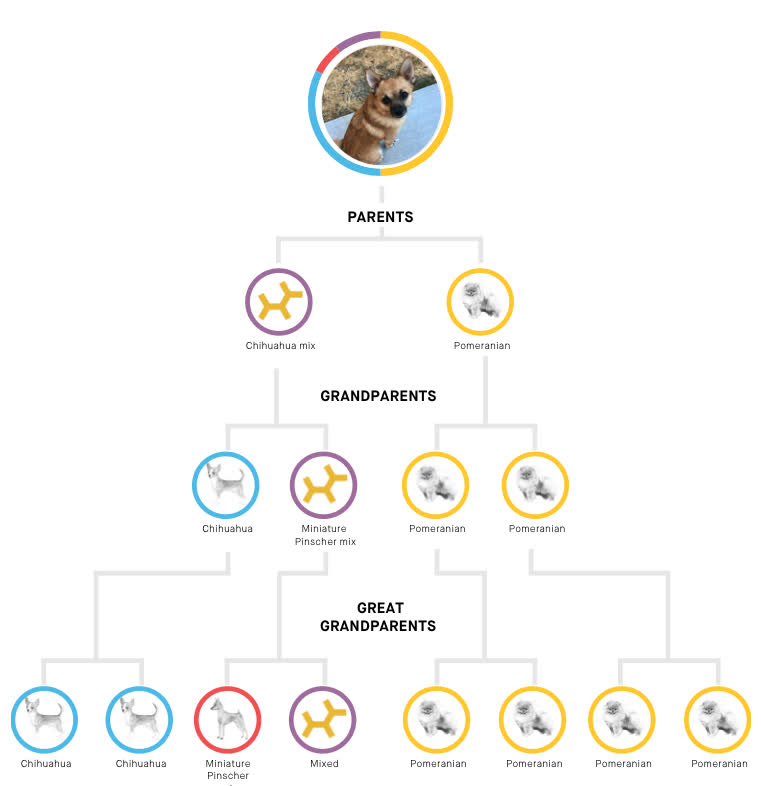
This means Riker's parents were a chihuahua mix and a purebred pomeranian.
Analyzing my dog's DNA
Besides finally being able to see my dog's genetic history, I was super happy that I could download the raw genetic data. When I unzipped the file, I was left with two text files:
riker.tfam // 75B
riker.tped // 6.7MB
(I renamed them for brevity)
I searched for information on these file types. They both seem to be related to the free PLINK genetics software package.
If we download PLINK and run it on these files, we get:
$ plink --tfile riker
Processing .tped file... 77%
Error: Invalid chromosome code '27' on line 166046 of .tped file.
(This is disallowed for humans. Check if the problem is with your data, or if
you forgot to define a different chromosome set with e.g. --chr-set.)
This is pretty cool! PLINK defaults to human DNA. This file is from a dog, not a person. Looking through the plink --help file, we can see that they have support for lots of species:
--cow/--dog/--horse/--mouse/--rice/--sheep : Shortcuts for those species.
(I wonder why rice is so interesting to the software...)
Anyways, let's run it with the --dog flag:
$ plink --dog --tfile riker
Processing .tped file... done.
plink.bed + plink.bim + plink.fam written.
PLINK wrote these files to the same directory:
$ ls
plink.bed
plink.bim
plink.fam
plink.log
riker.tfam
riker.tped
These look to be PLINK metadata files that the software uses to do its processing.
We can use PLINK to do some interesting sounding genetic computations. For example, if we run it with the --homozyg flag, we can see homozygosity reports. According to Wikipedia (I must have been in Johto during this part of biology), zygosity is "the degree of similarity of the alleles for a trait in an organism". Running it produces:
$ plink --dog --tfile riker --homozyg
1 dog (0 males, 1 female) loaded from .fam.
--homozyg: Scan complete, found 27 ROH.
Results saved to plink.hom + plink.hom.indiv + plink.hom.summary .
The software knows that the dog is female, which is pretty cool. The files it generated seem to indicate the degree of homozygosity for her individual genes. Neat!
If we run with the --het flag, we can see inbreeding coefficients. The file it produces show this:
O(HOM) E(HOM) N(NM) F
0 3.051e+04 61022 -1
From this helpful documentation I found online:
O(HOM) Observed number of homozygotes
E(HOM) Expected number of homozygotes
N(NM) Number of non-missing genotypes
F F inbreeding coefficient estimate
-1 looks like it indicates a sampling error or contamination according to the docs:
The estimate of F can sometimes be negative. Often this will just reflect random sampling error, but a result that is strongly negative (i.e. an individual has fewer homozygotes than one would expect by chance at the genome-wide level) can reflect other factors, e.g. sample contamination events perhaps.
We can also use the software to find what parts of the genotyping are missing with the --missing flag. From this, I was able to gather that Riker only has a missing SNP rate of 0.0009034 (less than 1%). I think this means that Riker's DNA in this sample is over 99% complete. Cool!
I may make Riker's DNA available online some day for others to do genetic analysis.
Anyways, I thought this was a fun exercise into how genetic data is stored and processed. It's really cool that there is open source software to analyze this data. Thanks for reading!